
In the last article, I already finished A and B part of the biotechnology chapter. In this section, I would finish the (C) part of the chapter.
(C) RECOMBINANT DNA TECHNOLOGY
- Isolation of DNA
- Fragmentation of DNA by specific RE
- Isolation of desired DNA fragment through agarose-gel electrophoresis
- Ligation of DNA fragment into a vector treated with similar RE
- Transferring the recombinant DNA into the host
- Culturing the host cells in a medium at large scale
- Extraction of the desired product
(A) ISOLATION OF DNA:
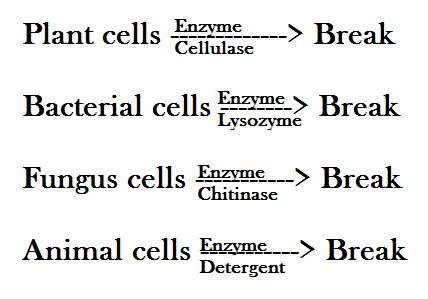
What could be the source of the genetic material to be used in the genetic engineering / recombinant DNA technology? Well, we can extract DNA from various sources such as plant cells, animal cells, bacterial cells and fungal cells, to name only a few.
Now, since DNA is present within the cell, the cell membrane including the nuclear membrane needs to be broken down to release DNA along with other macromolecules such as RNA, proteins, lipids and polysaccharides. This can be achieved using different enzymes for different cells.
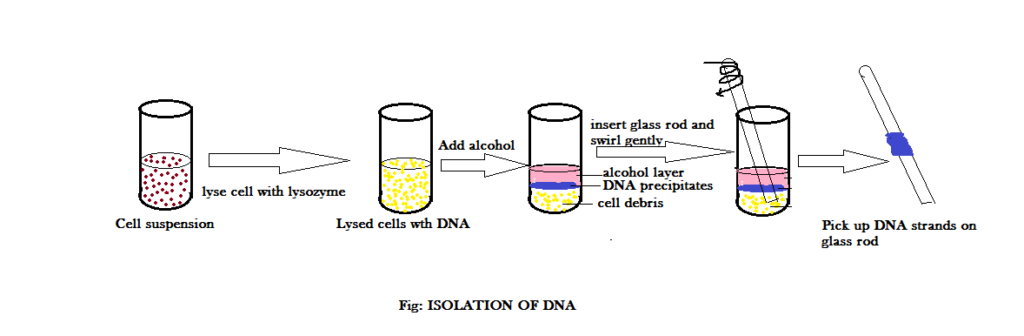
Once the cell is broken down, the cell content is treated with carbohydrases, proteases, lipases and RNAse to destroy these macromolecules. Then it is treated with chilled ethanol for the precipitation of DNA. Since DNA is not soluble in ethanol, it precipitates out. The precipitated DNA can be spooled on to a glass rod – this process is called SPOOLING (a method to remove DNA). The figure below demonstrates the complete procedure:
(B) Fragmentation of DNA by specific Restriction Endonuclease
In this process, of recombinant DNA technology, the isolated DNA (foreign DNA / passenger DNA) and the vector DNA is cut using the same restriction endonuclease enzyme. The resultant DNA fragments would have the same sticky ends (if the restriction enzyme is say, E. coli).
(C) Isolation of desired DNA fragment through agarose-gel electrophoresis
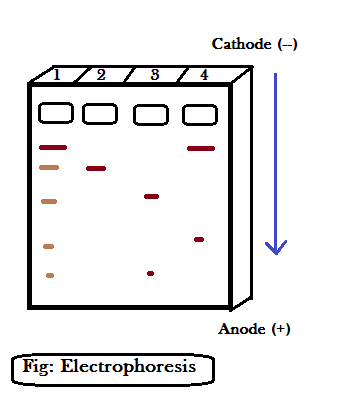
Gel electrophoresis is a method through which DNA fragments are separated based on their sizes. The most commonly used matrix in gel electrophoresis is agarose, which is a natural polymer extracted from sea weeds (algae).
The DNA fragments separate according to their size through sieving effect provided by the agarose gel. Thus, the smaller fragments move farther than the large one.
All the DNA fragments collected after using the restriction endonuclease are poured into the well (1,2,3,4) present in the device shown above. The larger DNA fragments move slowly than the smaller DNA fragments in the agarose gel.
Once DNA fragments are separated – nothing can be seen in the gel unless the gel is stained with ETHIDIUM BROMIDE followed by exposure to the UV radiation. Once the gel is exposed to the UV radiation, DNA bands are seen, which appear orange in color. The DNA bands containing agarose-gel are cut out in the form of gel pieces. The DNA (gene of interest) containing gel is cut out from the whole block of agar – this procedure is known as ELUTION. Now, you can ask a question, how do you know which DNA fragment is our gene of interest? Well, we know beforehand the size of our DNA fragment (gene of interest). In the electrophoresis device, there is a measured DNA band, say, in the first well (1). Based on the measured bands, we can see the length of each DNA band on the other wells and thus extract the DNA band of interest.
Once the DNA (gene of interest) has been removed from the gel, it needs to be amplified to make several copies of it. This is done through POLYMERASE CHAIN REACTION (PCR).
POLYMERASE CHAIN REACTION (PCR)
PCR (Polymerase Chain Reaction) technology was developed by Kerry Mullis in 1985. It is a technique through which selective amplification of a specific region of DNA molecule is performed. Now, the question is why PCR is required? Well, if you have a single DNA fragment (gene of interest) and using that DNA you’re performing an experiment – there is a maximum chance that the DNA fragment would get damaged while working with it. It is therefore necessary to have multiple copies of the DNA fragments to avoid any kind of damage to the DNA (gene of interest).
Principle behind PCR Technology:
The basic principle behind PCR is that when a double stranded DNA is heated, the DNA strands separate giving rise to single stranded molecules, which can be used to hybridize with small oligonucleotide primers (single stranded) by bridging down the molecule. DNA polymerase and nucleotide triphosphates are added to continue the process of replication on the primer site. This procedure is repeated several times, which finally results in the amplification of DNA between the two primers (one on each strand of separated DNA).
Basic Requirements to Perform PCR
- DNA template to be amplified
- Primers, which is an oligonucleotide, usually 10-18 bp long
- DNA polymerase, which is stable at temperature above 80 degree Celsius. Taq polymerase which has been isolated from a thermostable bacterial species (Thermus aquaticus) is used.
- Deoxynucleotide triphosphates and buffer
Steps of PCR:
- Denaturation: The target sequence of DNA is heated to denature the template strands so that the two strands get separated.
- Annealing: The DNA is cooled to allow the process to anneal i.e., the primers to attach to both the separated DNA strands.
- Extension: In the presence of Mg ion, DNA polymerase extends the primers on both strands from 5’-3’ direction.

(D) Ligation of DNA fragment into a vector treated with similar RE:
When DNA is isolated from a cell, it is treated with restriction endonuclease enzyme that cuts DNA into fragments. All fragments are separated through gel electrophoresis on the basis of their respective lengths and the desired DNA (gene of interest) is isolated. On the other hand, the vector DNA is also cut with the same restriction endonuclease to cut its DNA. Now the desired DNA (passenger DNA) is attached to the vector DNA to form a recombinant DNA. The desired DNA segment and vector DNA is attached with the help of the enzyme, called DNA LiGASE.
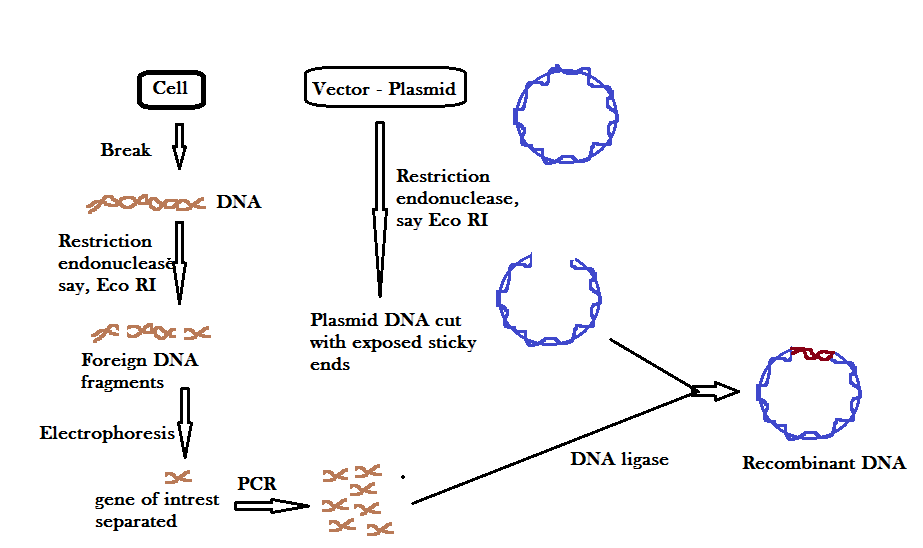
Note: the attachment of a foreign DNA is carried out at a specific restriction site present in the antibiotic resistance gene (selectable marker). For example, if we attach a foreign DNA at Bam H1 site of tetracycline resistance gene in the vector pBR322 – the plasmids will lose tetracycline resistance, but can still be selected out from non-recombinant ones by plating the transformants on ampicillin containing medium. The inactivation of selectable marker gene by replacing it with a foreign gene is called INSERTIONAL INACTIVATION.
The first artificial recombinant DNA was formed by Herbert and Stanley in 1972. They incorporated a DNA sequence (gene encoding antibiotic resistance) with a native plasmid, Salmonella typhimurium.
(E) Transferring the recombinant DNA into the host:
After a recombinant DNA molecule has been formed, it is introduced into the suitable host. There are various methods through which recombinant DNA is introduced into the host cell depending on several factors including the vector type and the host cell.
(a) TRANSFORMATION:
this is considered as the most common method of introducing rDNA into the living cells. In this process, bacterial cells take up rDNA from the surrounding environment. However, many host cells such as E. coli, yeast and mammalian cells do not readily take up foreign DNA and thus they are chemically treated to become competent. For instance, E. coli is first treated with CaCl2 solution where CaCl2 breaks into ions as Ca2+ and Cl-. The divalent cation Ca2+ increases the efficiency with which DNA enters the bacterium through pores in the bacterial cell wall. After that the rDNA is incubated on ice, followed by placing them briefly at 42 degree Celsius (heat shock) and then putting them back on ice. This enables the bacteria to take up the rDNA.
(b) TRANSFECTION:
it is a method of transfer of rDNA into eukaryotic cells. In this procedure, the rDNA is mixed with charged substances like cationic liposome, calcium phosphate, etc and overlaying on recipient host cell, host cell takes up the rDNA.
(c) Direct (vectorless) gene transfer:
- Microinjection
- Chemical-mediated gene transfer
- Biolistics or gene-gun method
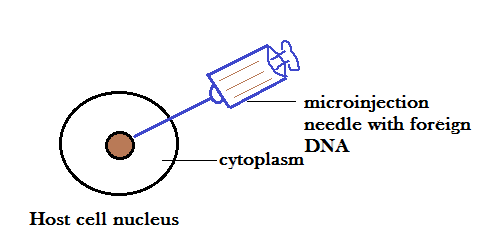
MICROINJECTION:
In this method, foreign DNA is directly injected into the nucleus of the cell using microneedle or micropippete. This method is generally used for the microinjection of eggs, oocytes and embryos.
CELL MEDIATED GENE TRANSFER
Certain chemicals like PEG (Polyethylene glycol) help foreign DNA to enter into the host cell.
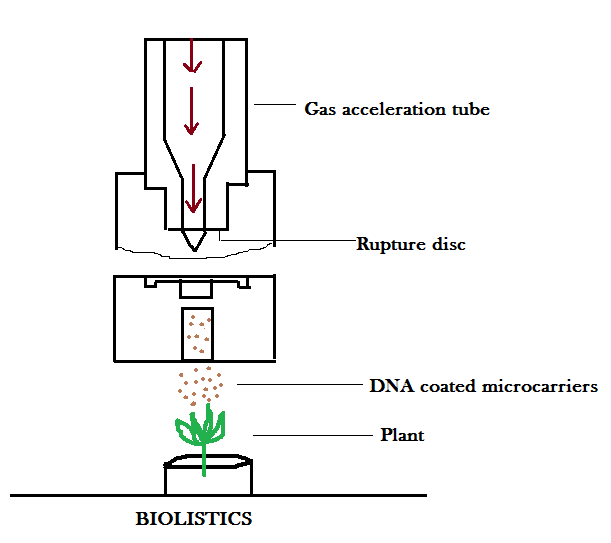
BIOLISTICS
This method is suitable for plant cells having call wall. In this method, plant cells are bombarded with high velocity micro particles of gold or tungsten coated with DNA.
Host Cell with Recombinant DNA Containing Vector Possibilities:
When a recombinant vector is introduced in a host bacterium, say, E.coli – there could be three possibilities: 1. the cloning vector could not enter into the host cell – such host cell is called NON-TRANSFORMANT CELL. 2. The vector successfully introduced into the host cell – such cell is called TRANSFORMANT CELL. This can further be divided into two categories: non-recombinant transformant and recombinant transformant. Non-recombinant transformants are those in which the vector introduced successfully, but that vector is non-recombinant meaning without the foreign gene or without insertional inactivation. And the recombinant transformant is the one in which the recombinant vector is present meaning insertional inactivation is present due to the presence of foreign / passenger DNA.
How to Select Transformants?
1. The host cell will be transferred to the medium containing ampicillin:
In such a medium, non-transformants (host without recombinant DNA containing vector) will not survive. Only transformants will survive in such a medium. But the question is transformants are also of two types: non-recombinant and recombinant. The next step would be to separate the two.
2. Separation of recombinant and non-recombinant transformants:
Transfer the cells into the medium containing tetracycline – if the host survives, it means, it is a non-recombinant and if sensitive, it is recombinant. However, when both non-recombinant and recombinant are placed in ampicillin culture, both will survive and form colonies. The culture media plate is called MASTER PLATE.
Replica Plating Experiment to Separate Recombinant from Non-recombinant:
Using a valveteen pad (replicator) a replica plate is formed. This replica plate is not put on a tetracycline culture media. Now, we compare replica plate with master plate, and we can clearly spot recombinant from non-recombinant. We then extract recombinant from the master plate and culture the same.
Well, the above procedure to separate recombinants from non-recombinants is a cumbersome procedure and thus an alternative method has been developed to separate the two. An alternative selective marker has been developed, which differentiate recombinants from non recombinants on the basis of their ability to produce color in the presence of a CHROMOGENIC SUBSTRATE.
Steps: PAGE- 200 NCERT
- The selectable marker for the enzyme galactosidase is developed and inserted into the vector DNA
- A recombinant DNA is inserted within the coding sequence of the enzyme galactosidase, which results in the inactivation of the enzyme.
- The presence of chromogenic substrate gives blue colored colonies if the plasmid of the bacteria does not have an insert.
- If the insert is present in the plasmid DNA at the coding sequence of the alpha-galactosidase enzyme – the colonies do not produce any color – they are identified as recombinant colonies.
(F) Culturing the host cells in a medium at large scale:
Once the recombinants (transformed cells) are collected – they are place in a suitable medium where each transformed cell forms a colony of identical cells, often called a CLONE. Now, this clone is utilized for the production of desirable products. This can be achieved at three steps:
(i) Laboratory Scale Process
(ii) Pilot Plant Scale
(iii) Manufacturing unit
LABORATORY SCALE:
In this process, the clones are selected and multiplied. Proper medium is also selected in which best product and more amount of product could be formed. All experiments performed in the lab include GLASS APPARATUS. The laboratory scale processes are finalized and transferred at Pilot-plant scale.
PILOT PLANT SCALE:
It is the intermediate stage where working of laboratory scale process is tested. At this stage, cost and quality of product is thoroughly checked. Glass apparatus is replaced by stainless steel equipment – called BIOREACTORS.
Bioreactors are used to produce 100-1000 liters of products such as enzymes and proteins. You can think of a bioreactors as vessels in which raw materials are biologically converted into specific products, enzymes, etc using microbial plant, animal or human cells.
The most commonly used bioreactors are Simple Stirred Tank Bioreactors.
Simple Stirred Tank Bioreactors (continuous production of products): this bioreactor is made up of stainless steel body. This bioreactor can be separated into two regions:
- Working volume: It comprises of 70-80% of the total volume consisting of medium, microbes and gas bubbles.
- Head-space volume: It comprises of 20-30% of the total volume.

During the designing of a bioreactor – often large size is considered to accommodate large amount of the culture medium.
Broth: nutrient medium
A bioreactor has an agitator system, oxygen delivery system and a foam control system, a temperature control system, pH control system and sampling parts so that small volumes of culture can be withdrawn periodically. There are two types of systems involved in the manufacturing of products:
- Upstream processing: It involves continuous production of products. In other words, products are made continuously. It includes all the processes up to the formation of recombinant product or transgenic product.
- Downstream processing: After completion of the manufacturing stage, the products need to be subjected through a series of processes before it is ready for marketing as a finished product. The downstream processing includes:
(i) Isolation
(ii) Purification
(iii) Purity testing
(iv) Addition of preservatives, if any
(v) Packaging for marketing
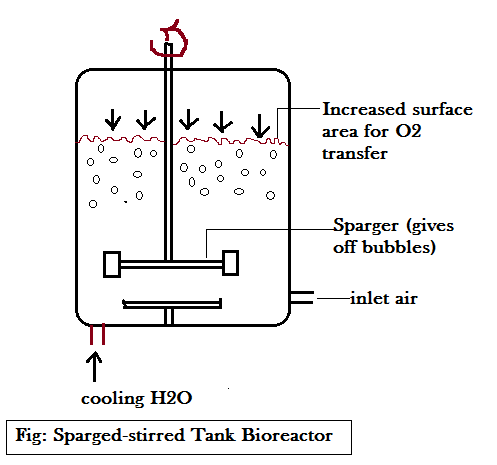
Sparged-stirred Tank Bioreactor (discontinuous process): This bioreactor is discontinuous type meaning products are removed, raw materials are added sequentially.
Frequently Asked Questions (FAQs)
Q. How can we determine whether the plasmid’s genetic information will actually be expressed inside the bacteria?
A. Plasmid expression in bacteria is confirmed using marker genes and screening tests. For example, antibiotic resistance shows plasmid uptake, while color change (like blue-white screening) confirms gene expression.
Q. After transformation, how can we confirm that the insert is integrated into the host genome? Aside from using GFP fluorescence, what other simple methods can verify this — or is insertion always guaranteed?
A. Insertion into the host genome isn’t guaranteed, so confirmation is essential. Besides using GFP fluorescence, common methods include PCR to detect the insert’s DNA sequence, restriction digestion to check fragment patterns, and sequencing to verify correct integration. The simplest and most routine method is colony PCR, which quickly confirms whether the insert is present in transformed cells.
Q. How are PCR primers made with such precise, tiny nucleotide sequences — and how are the specific enzymes produced as well?
A. PCR primers are made through solid-phase chemical synthesis, where nucleotides are added one by one to a chain fixed on a solid support. Each step ensures accuracy through coupling, capping, and oxidation, followed by HPLC purification and quality checks.
Enzymes like DNA polymerases are produced using recombinant DNA technology — the enzyme’s gene is inserted into host cells (like E. coli), which are grown to express the enzyme. It’s then purified and tested for precision and reliability.
Q. In PCR, why is the temperature lowered during the primer-binding step?
A. The cooling step in PCR allows primers to bind (anneal) to their complementary sequences on the single-stranded DNA templates. At high temperatures, DNA strands remain separated, but lowering the temperature enables hydrogen bonds to form between primers and the target DNA, ensuring accurate primer attachment before extension begins.